SAR of New Hetero-Monocyclic Analogs of (-)-Zampanolide
The marine natural product zampanolide (1) acts as a potent microtubule inhibitor with binding at the taxane site on b-tubulin and it inhibits human cancer cell proliferation in vitro with single digit nM IC50 values. The structure of zampanolide (1) incorporates a highly unsaturated 20-membered macrolactone ring, an N-acyl hemiaminal-linked side chain, and a THP ring containing an exo-methylene group.1
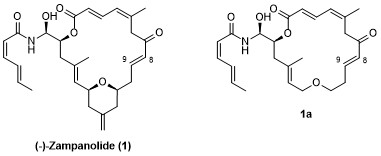
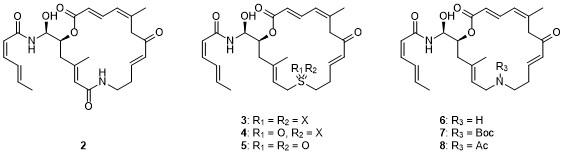
Zampanolide binds to b-tubulin in a covalent fashion through 1,4-addition of His229 to the C8 – C9 double bond in the macrocycle. In contrast to the essential nature of the enone double bond, we have previously shown that the removal of the THP ring in zampanolide (1) is relatively well tolerated; thus, des-THP analog 1a retains significant antiproliferative activity.2 As part of a comprehensive project on the SAR of zampanolide-type structures, we have targeted various heteroatom-analogs of 1a for synthesis (2-8), in order to evaluate if some the activity difference between 1 and 1a could be recovered without a (re)increase in structural complexity.3
This contribution will discuss the synthetic chemistry of zampanolide analogs 2-8. In addition, the biological activity of selected compounds will be presented.
[1] Jun-Ichi Tanaka, Tatsuo Higa, Tetrahedron Letters, 1996, 37, 5535–5538.
[2] Didier Zurwerra, Florian Glaus, Leo Betschart, Julia Schuster, Jürg Gertsch, Walter Ganci, Karl-Heinz Altmann, Chemistry - A European Journal, 2012, 18, 16868–16883.
[3] Andrea E. Prota, Katja Bargsten, Didier Zurwerra, Jessica J. Field, José F. Diaz, Karl-Heinz Altmann, Michel O. Steinmetz, Science, 2013, 339, 587–590.