Solvent Relaxation and its Influence on Photochemical Reactions
The choice of the solvent can have a dramatic effect on the outcome of a chemical reaction. In addition to specific solvent interactions such as hydrogen bonding, solvent environment can greatly influence equilibrium, reaction rate and driving force by different stabilization of the reactants, transition states or products, especially with reactions involving charged or strongly dipolar species. For example, the activation energy is decreased when solvation energy of the transition state is larger than that of the reactant state, whereas the overall driving force of a reaction is increased upon greater stabilization of the product state relative to the reactant state.[1] All these above effects can be considered as equilibrium effects and can be illustrated by a static energy-level diagram. How about when the rate of a solvent influenced reaction is faster or comparable to the dynamics of the solvent itself? In this case, the situation cannot be illustrated by a static picture and the reaction becomes solvent controlled i.e. both the rate and the yield of the reaction are controlled by the solvent relaxation.[2]
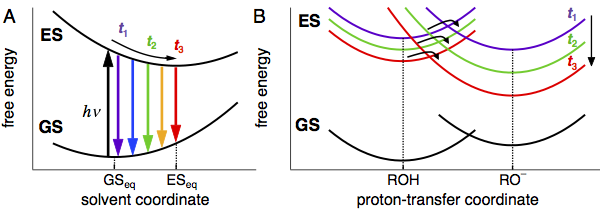
In our communication, we will explain the basic principles of dynamic solvent relaxation: What is its origin and how can we monitor this process?[3] Second, we will demonstrate the effect of solvent relaxation on a photochemical reaction, namely an excited-state proton transfer (ESPT). This reaction consists of the photodissociation of a hydroxylic proton from an aromatic alcohol and results in formation of charged products. The reaction is therefore strongly influenced by the solvent environment making it an ideal candidate to study the dynamic solvent effects.
[1] Christian Reichardt, Thomas Welton (2010) Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, Germany, pp. 107–357.
[2] T. Kumpulainen, B. Lang, A. Rosspeintner, E. Vauthey, Chem. Rev., 2016, DOI: 10.1021/acs.chemrev.6b00491.
[3] T. Kumpulainen, A. Rosspeintner, E. Vauthey, Phys. Chem. Chem. Phys., 2017, 19, 8815-8825.